

中華人民共和國國家標準
煤的元素分析方法GB476—91
代替GB476—79
Ultimate analysis of coal
國家技術監督局1991-05-22 批準1992-03-01 實施
本標準參照采用了國際標準1SO625:1975(E)《煤和焦炭碳和氫測定方法利比西法》和ISO 333:1983(E)《煤氮測定方法半微量開氏法》。
1 主題內容與適用范圍
本標準規定了煤中碳、氫、氮含量的測定方法和氧含量的計算方法。
本標準適用于褐煤、煙煤和無煙煤。
2 引用標準
GB211 煤中全水分的測定方法
GB212 煤的工業分析方法
GB214 煤中全硫的測定方法
GB218 煤中碳酸鹽二氧化碳含量的測定方法
3 碳和氫的測定
3.1 方法提要
稱取一定量的空氣干燥煤樣在氧氣流中燃燒,生成的水和二氧化碳分別用吸水劑和二氧化碳吸收劑吸收,由吸收劑的增重計算煤中碳和氫的含量。煤樣中硫和氯對測定的干擾在三節爐中用鉻酸鉛和銀絲卷消除,在二節爐中用高錳酸銀熱解產物消除。氮對碳測定的干擾用粒狀二氧化錳消除。
3.2 試劑和材料
3.2.1 堿石棉:化學純,粒度1~2 mm;或堿石灰(HGB3213):化學純,粒度0.5~2mm。
3.2.2 無水氯化鈣(HGB3208):分析純,粒度2~5mm;或無水過氯酸鎂;分析純,粒度1~3mm。
3.2.3 氧化銅(HGB3438):分析純,粒度1~4mm,或線狀(長約5mm)。
3.2.4 鉻酸鉛(HG3—1071):分析純,粒度1~4mm。
3.2.5 銀絲卷:絲直徑約0.25 mm。
3.2.6 銅絲卷:絲直徑約0.5 mm。
3.2.7 氧氣:不含氫。
3.2.8 三氧化二鉻(HG 3—933):化學純,粉狀,或由重鉻酸銨、鉻酸銨加熱分解制成。
制法:取少量鉻酸銨放在較大的蒸發皿中,微微加熱,銨鹽立即分解成墨綠色、疏松狀的三氧化二鉻。收集后放在馬弗爐中,在600±10℃下灼燒40min,放在空氣中使呈空氣干燥狀態,保存在密閉容器中備用。
3.2.9 粒狀二氧化錳:用化學純硫酸錳(HG 3—1081)和化學純高錳酸鉀(GB643)制備。
制法:稱取25g 硫酸錳(MnSO4·5H2O),溶于500mL 蒸餾水中,另稱取16.4g 高錳酸鉀,溶于300mL 蒸餾水中,分別加熱到50~60℃。然后將高錳酸鉀溶液慢慢注入硫酸錳溶液中,并加以劇烈攪拌。之后加入10mL、(1+1)硫酸(GB625,化學純),將溶液加熱到70~80℃并繼續攪拌5min,停止加熱,靜置2~3h。用熱蒸餾水以傾瀉法洗至中性,將沉淀物移至漏斗過濾,然后放入干燥箱中,在150℃左右干燥,得到褐色、疏松狀的二氧化錳,小心破碎和過篩,取粒度0.5~2mm 的備用。
3.2.10 氧化氮指示膠:
制法: 在瓷蒸發皿中將粒度小于2 mm 的無色硅膠40g 和濃鹽酸30mL 攪拌均勻。在沙浴上把多余的鹽酸蒸干至看不到明顯的蒸氣逸出為止。然后把硅膠粒浸入30mL、10%硫酸氫鉀溶液中,攪拌均勻取出干燥。再將它浸入30mL、0.2%的雷伏奴耳(乳酸-6,9-二氨基-2-乙氧基吖啶)溶液中,攪拌均勻,用黑色紙包好干燥,放在深色瓶中,置于暗處保存,備用。
3.2.11 高錳酸銀熱解產物:當使用二節爐時,需制備高錳酸銀熱解產物。
制法:稱取100g 化學純高錳酸鉀(GB643),溶于2L 蒸餾水中,另取107.5g 化學純硝酸銀(GB670)先溶于約50mL 蒸餾水中,在不斷攪拌下,傾入沸騰的高錳酸鉀溶液中。攪拌均勻,逐漸冷卻,靜置過夜。將生成的具有光澤的、深紫色晶體用蒸餾水洗滌數次。在60~80℃下干燥4h。將晶體一點一點地放在瓷皿中,在電爐上緩緩加熱至驟然分解,得疏松狀、銀灰色產物,收集在磨口瓶中備用。未分解的高錳酸鉀不宜大量貯存,以免受熱分解,不安全。
3.3 儀器、設備
3.3.1 碳氫測定儀
碳氫測定儀包括凈化系統、燃燒裝置和吸收系統三個主要部分,結構如圖1 所示。
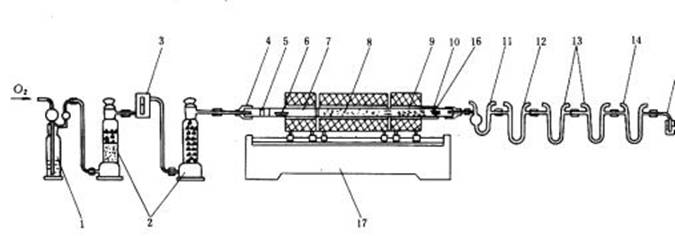
圖1 碳氫測定儀
1—鵝頭洗氣瓶;2—氣體干燥塔;3—流量計;4—橡皮帽;5—銅絲卷;6—燃燒舟;7—燃燒管;8—氧化銅;9—鉻酸鉛;10—銀絲卷;11—吸水U 形管;12—除氮U 形管;13—吸二氧化碳U 形管;14—保護用U 形管;15—氣泡計;16—保溫套管;17—三節電爐
3.3.1.1 凈化系統:包括以下部件:
a.鵝頭洗氣瓶:容量250~500mL,內裝40%氫氧化鉀(或氫氧化鈉)溶液;
b.氣體干燥塔:容量500mL2 個,一個上部(約2/3)裝氯化鈣(或過氯酸鎂),下部(約1/3)裝堿石棉(或堿石灰);另一個裝氯化鈣(或過氯酸鎂);
c.流量計:量程0~150mL/min。
3.3.1.2 燃燒裝置:由一個三節(或二節)管式爐及其控制系統構成,主要包括以下部件:
a.電爐:三節爐或二節爐(包括雙管爐或單管爐),爐膛直徑約35mm;
三節爐:第一節長約230mm,可加熱到800±10℃并可沿水平方向移動;第二節長330~350mm,可加熱到800±10℃;第三節長130~150mm,可加熱到600±10℃。
二節爐:第一節長約230mm,可加熱到800±10℃并可沿水平方向移動;第二節長130~150 mm,可加熱到500±10℃。
每節爐裝有熱電偶,測溫和控溫裝置。
b.燃燒管:瓷、石英、剛玉或不銹鋼制成,長1100~1200mm(使用二節爐時,長約800mm),內徑20~22mm,壁厚約2 mm;
c.燃燒舟:瓷或石英制成,長約80mm;
d.保溫套:銅管或鐵管,長約150mm,內徑大于燃燒管,外徑小于爐膛直徑:
e.橡皮帽(最好用耐熱硅橡膠)或銅接頭。
3.3.1.3 吸收系統:包括以下部件:
a.吸水U 形管:如圖2 所示,裝藥部分高100~120mm,直徑約15mm,進口端有一個球形擴大部分,內裝無水氯化鈣或無水過氯酸鎂。
b.二氧化碳吸收管:2 個,如圖3 所示。裝藥部分高100~120mm,直徑約15mm,前2/3 裝堿石棉或堿石灰,后1/3 裝無水氯化鈣或無水過氯酸鎂。
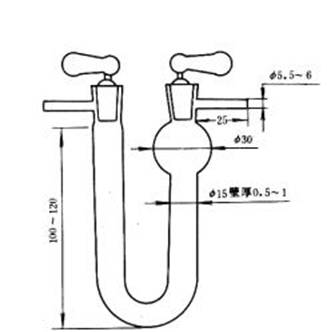
圖2 吸水U 形管
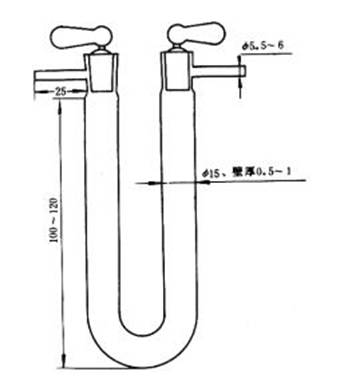
圖3 二氧化碳吸收管(或除氮U 形管)
c.除氮U 形管:如圖3 所示。裝藥部分高100~120mm,直徑約15mm,前2/3 裝二氧化錳,后1/3 裝無水氯化鈣或無水過氯酸鎂。
d.氣泡計:容量約10 mL。
3.3.2 分析天平:感量0.0001 g。
3.3.3 貯氣桶:容量不小于10 L。
3.3.4 下口瓶:容量約10 L。
3.3.5 帶磨口塞的玻璃管或小型干燥器(不裝干燥劑)。
3.4 試驗準備
3.4.1 凈化系統各容器的充填和連接
在3.3.1.1 條所述凈化系統各容器中裝入相應的凈化劑,然后按圖1 順序將各容器連接好。
氧氣可采用儲氣桶和下口瓶或可控制流速的氧氣瓶供給。為指示流速,在兩個干燥塔之間接入一個流量計。凈化劑經70~100 次測定后,應進行檢查或更換。
3.4.2 吸收系統各容器的充填和連接
在3.3.1.3 條所述吸收系統各容器中裝入相應的吸收劑,然后按圖1 順序將各容器連接好。吸收系統的末端可連接一個空U 形管(防止硫酸倒吸)和一個裝有硫酸的氣泡計。如果作吸水劑用的氯化鈣含有堿性物質,應先以二氧化碳飽和。然后除去過剩的二氧化碳。處理方法如下:
把無水氯化鈣破碎至需要的粒度(如果氯化鈣在保存和破碎中已吸水,可放入馬弗爐中在約300℃下灼燒1h)裝入干燥塔或其他適當的容器內(每次串聯若干個)。緩慢通入干燥的二氧化碳氣3~4h,然后關閉干燥塔,放置過夜。通入不含二氧化碳的干燥空氣,將過剩的二氧化碳除盡。處理后的氯化鈣貯于密閉的容器中備用。當出現下列現象時,應更換U 形管中試劑:
a.U 形管中的氯化鈣開始溶化并阻礙氣體暢通;
b.第二個吸收二氧化碳的U 形管做一次試驗、其質量增加達50 mg 時,應更換第一個U 形管中的二氧化碳吸收劑;
c.二氧化錳一般使用50 次左右應進行檢查或更換。
檢查方法:將氧化氮指示膠裝在玻璃管中,兩端堵以棉花,接在除氮管后面。或將指示膠少許放在二氧化碳吸收管進氣端棉花處。燃燒煤樣,若指示膠由草綠色變成血紅色,表示應更換二氧化錳。
上述U 形管更換試劑后,通入氧氣待質量恒定后方能使用。
3.4.3 燃燒管的填充
使用三節爐時,按圖4 填充:
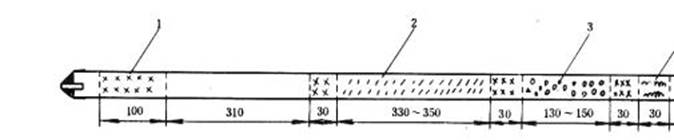
圖4 三節爐燃燒管填充示意圖
1—銅絲卷;2—氧化銅;3—鉻酸鉛;4—銀絲卷
首先制做三個長約30 mm 和一個長約100 mm 的絲直徑約0.5mm 銅絲卷,直徑稍小于燃燒管的內徑,使之既能自由插入管內又與管壁密接。制成的銅絲卷應在馬弗爐中于800℃左右灼燒1h 后再用。
燃燒管出氣端留50mm 空間,然后依次充填30 mm 絲直徑約0.25mm 銀絲卷,30mm銅絲卷,130~150mm(與第三節電爐長度相等)鉻酸鉛(使用石英管時,應用銅片把鉻酸鉛與管隔開),30mm 銅絲卷,330~350mm(與第二節電爐長度相等)粒狀或線狀氧化銅,30mm銅絲卷,310mm 空間(與第一節電爐上燃燒舟長度相等)和100mm 銅絲卷。燃燒管兩端裝以橡皮帽或銅接頭,以便分別同凈化系統和吸收系統連接。橡皮帽使用前應預先在105~110℃下干燥8h 左右。
燃燒管中的填充物(氧化銅、鉻酸鉛和銀絲卷)經70~100 次測定后應檢查或更換1) 。
注:1) 下列幾種填充劑經處理后可重復使用:
氧化銅用1mm 孔徑篩子篩去粉末,篩上的氧化銅備用;鉻酸鉛可用熱的稀堿液(約5%氫氧化鈉溶液)浸漬,用水洗凈、干燥,并在500~600
℃下灼燒0.5h 以上后使用;銀絲卷用濃氨水浸泡5min,在蒸餾水中煮沸5min,用蒸餾水沖洗干凈,干燥后再用。
使用二節爐時按圖5 填充:
首先制成兩個長約10mm 和一個長約100 mm 的銅絲卷,再用3~4 層100 目銅絲布剪成的圓形墊片與燃燒管密接,用以防止粉狀高錳酸銀熱解產物被氧氣流帶出,然后按圖5裝好。
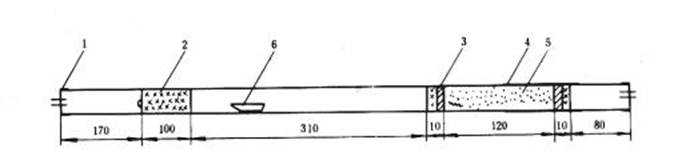
圖5 二節爐燃燒管填充示意圖
1—橡皮帽;2—銅絲卷;3—銅絲布圓墊;4—保溫套管;5—高錳酸銀熱解產物;6—瓷舟
3.4.4 爐溫的校正
將工作熱電偶插入三節爐的熱電偶孔內,使熱端稍進入爐膛,熱電偶與高溫計連接。將爐溫升至規定溫度,保溫1h。然后將標準熱電偶依次插到空燃燒管中對應于第一、第二、第三節爐的中心處(注意勿使熱電偶和燃燒管管壁接觸)。調節電壓,使標準熱電偶達到規定溫度并恒溫5min。記下工作熱電偶相應的讀數,以后即以此為準控制溫度。
3.4.5 空白試驗
將裝置按圖1 連接好,檢查整個系統的氣密性,直到每一部分都不漏氣以后,開始通電升溫,并接通氧氣。在升溫過程中,將第一節電爐往返移動幾次,并將新裝好的吸收系統通氣20min 左右。取下吸收系統,用絨布擦凈,在天平旁放置10min 左右,稱量。當第一節和第二節爐達到并保持在800±10℃,第三節爐達到并保持在600±10℃后開始作空白試驗。此時將第一節爐移至緊靠第二節爐,接上已經通氣并稱量過的吸收系統。在一個燃燒舟上加入氧化鉻(數量和煤樣分析時相當)。打開橡皮帽,取出銅絲卷,將裝有氧化鉻的燃燒舟用鎳鉻絲推至第一節爐入口處,將銅絲卷放在燃燒舟后面,套緊橡皮帽,接通氧氣,調節氧氣流量為120mL/min。移動第一節爐,使燃燒舟位于爐子中心。通氣23min,將爐子移回原位。2min 后取下U 形管,用絨布擦凈,在天平旁放置10min 后稱量。吸水U 形管的質量增加數即為空白值。重復上述試驗,直到連續兩次所得空白值相差不超過0.0010g,除氮管、二氧化碳吸收管最后一次質量變化不超過0.0005g 為止。取兩次空白值的平均值作為當天氫的空白值。
在做空白試驗前,應先確定保溫套管的位置,使出口端溫度盡可能高又不會使橡皮帽熱分解。如空白值不易達到穩定,則可適當調節保溫管的位置。
3.5 分析步驟
3.5.1 將第一節和第二節爐溫控制在800±10℃,第三節爐溫控制在600±10℃,并使第一節爐緊靠第二節爐。
3.5.2 在預先灼燒過的燃燒舟中稱取粒度小于0.2 mm 的空氣干燥煤樣0.2g,精確至0.0002g,并均勻鋪平。在煤樣上鋪一層三氧化二鉻。可把燃燒舟暫存入專用的磨口玻璃管或不加干燥劑的干燥器中。
3.5.3 接上已稱量的吸收系統,并以120mL/min 的流量通入氧氣。關閉靠近燃燒管出口端的U 形管,打開橡皮帽,取出銅絲卷,迅速將燃燒舟放入燃燒管中,使其前端剛好在第一節爐口。再將銅絲卷放在燃燒舟后面,套緊橡皮帽,立即開啟U 形管,通入氧氣,并保持120mL/min的流量。1min 后向凈化系統方向移動第一節爐,使燃燒舟的一半進入爐子。過2min,使燃燒舟全部進入爐子。再過2min,使燃燒舟位于爐子中心。保溫18min 后,把第一節爐移回原位。2min 后,停止排水抽氣。關閉和拆下吸收系統,用絨布擦凈,在天平旁放置10min后稱量(除氮管不稱量)。
3.5.4 也可使用二節爐進行碳、氫測定。此時第一節爐控溫在800±10℃,第二節爐控溫在500±10℃,并使第一節爐緊靠第二節爐。每次空白試驗時間為20min。燃燒舟位于爐子中心時,保溫13min,其他操作同第3.4.5、3.5.1、3.5.2 和3.5.3 條。
3.5.5 為了檢查測定裝置是否可靠,可稱取0.2~0.3g 分析純蔗糖(HG3—100)或分析純苯甲酸(HG3—987),加入20~30mg 純“硫華”進行3 次以上碳、氫測定。測定時,應先將試劑放入第一節爐爐口,再升溫,且移爐速度應放慢,以防標準有機試劑爆燃。如實測的碳、氫值與理論計算值的差值,氫不超過±0.10%,碳不超過±0.30%,并且無系統偏差,表明測定裝置可用,否則須查明原因并徹底糾正后才能進行正式測定。如使用二節爐,則在第一節爐移至緊靠第二節爐5min 以后,待爐口溫度降至100~200℃,再放有機試劑,并慢慢移爐,而不能采用上述降低爐溫的方法。
3.6 分析結果的計算
空氣干燥煤樣的碳氫含量按下式計算:
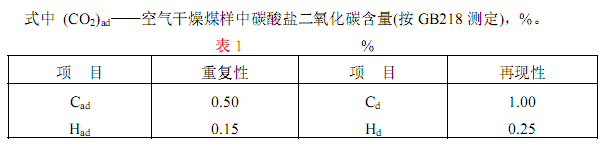
3.7 碳、氫測定的精密度碳、氫測定的重復性和再現性如表1 規定。
4 氮的測定
4.1 方法提要
稱取一定量的空氣干燥煤樣,加入混合催化劑和硫酸,加熱分解,氮轉化為硫酸氫銨。加入過量的氫氧化鈉溶液,把氨蒸出并吸收在硼酸溶液中,用硫酸標準溶液滴定。根據用去的硫酸量計算煤中氮的含量。
4.2 試劑
4.2.1 混合催化劑:將分析純無水硫酸鈉(HG3—123)32g、分析純硫酸汞5g 和分析純硒粉(HG3—926)0.5 g 研細,混合均勻備用。
4.2.2 鉻酸酐(HG 3—934):分析純。
4.2.3 硼酸(GB 628):分析純,3%水溶液,配制時加熱溶解并濾去不溶物。
4.2.4 混合堿溶液:將分析純氫氧化鈉(GB629)37g 和化學純硫化鈉(HG3—905)3g 溶解于蒸餾水中,配制成100mL 溶液。
4.2.5 甲基紅和亞甲基藍混合指示劑:
a.稱取0.175g 分析純甲基紅(HG3—958),研細,溶于50mL95%乙醇(GB679)中。
b.稱取0.083g 亞甲基藍(HGB 3364),溶于50mL95%乙醇(GB679)中。將溶液a 和b 分別存于棕色瓶中,用時按(1+1)混合。混合指示劑使用期不應超過1 周。
4.2.6 蔗糖(HG 3—1001):分析純。

![]()
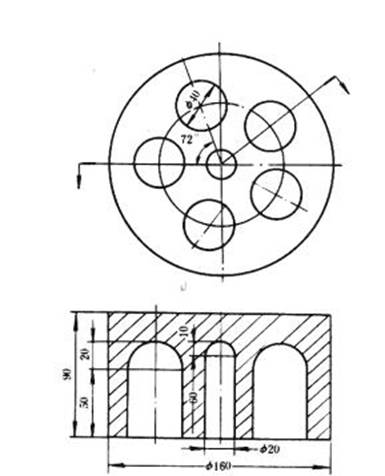
圖 6 鋁加熱體
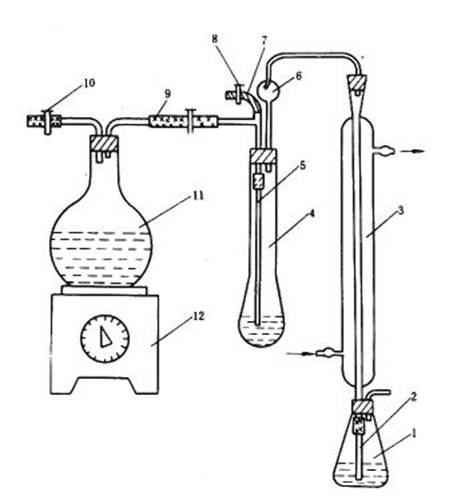
圖 7 蒸餾裝置
1—錐形瓶;2—橡皮管;3—直形玻璃冷凝管;4—開氏瓶;5—玻璃管;6—開氏球;7—橡皮管;
8—夾子;9、10—橡皮管和夾子;11—圓底燒瓶;12—萬能電爐
4.3 儀器、設備
4.3.1 開氏瓶:容量50mL 和250mL。
4.3.2 直形玻璃冷凝管:長約300mm。
4.3.3 短頸玻璃漏斗:直徑約30mm。
4.3.4 鋁加熱體:規格參照圖6,使用時四周圍以絕熱材料,如石棉繩等。
4.3.5 開氏球。
4.3.6 圓盤電爐:帶有調溫裝置。
4.3.7 錐形瓶:容量250mL。
4.3.8 圓底燒瓶:容量1000mL。
4.3.9 萬能電爐。
4.3.10 微量滴定管:10mL,分度值為0.05mL。
4.4 分析步驟
4.4.1 在薄紙上稱取粒度小于0.2mm 的空氣干燥煤樣0.2g,精確至0.0002g。把煤樣包好,放入50mL 開氏瓶中,加入混合催化劑2 g 和濃硫酸(相對密度1.84)5mL。然后將開氏瓶放入鋁加熱體的孔中,并用石棉板蓋住開氏瓶的球形部分。在瓶口插入一小漏斗,防止硒粉飛濺。在鋁加熱體中心的小孔中放溫度計。接通電源,緩緩加熱到350℃左右,保持此溫度,直到溶液清澈透明,漂浮的黑色顆粒完全消失為止。遇到分解不完全的煤樣時,可將0.2mm的空氣干燥煤樣磨細至0.1mm 以下,再按上述方法消化,但必須加入鉻酸酐0.2~0.5g。分解后如無黑色粒狀物且呈草綠色漿狀,表示消化完全。
4.4.2 將冷卻后的溶液,用少量蒸餾水稀釋后,移至250mL 開氏瓶中。充分洗凈原開氏瓶中的剩余物,使溶液體積約為100mL。然后將盛溶液的開氏瓶放在蒸餾裝置上準備蒸餾。蒸餾裝置如圖7 所示。
4.4.3 把直形玻璃冷凝管的上端連接到開氏球上,下端用橡皮管連上玻璃管,直接插入一個盛有20mL、3%硼酸溶液和1~2 滴混合指示劑的錐形瓶中。玻璃管浸入溶液并距離底約2mm。
4.4.4 在250mL 開氏瓶中注入25mL 混合堿溶液,然后通入蒸汽進行蒸餾,蒸餾至錐形瓶中溶液的總體積達到80mL 為止,此時硼酸溶液由紫色變成綠色。
4.4.5 蒸餾完畢后,拆下開氏瓶并停止供給蒸汽。插入硼酸溶液中的玻璃管內、外用蒸餾水沖洗。洗液收入錐形瓶中,用硫酸標準溶液滴定到溶液由綠色變成微紅色即為終點。由硫酸用量(校正空白)求出煤中氮的含量。
空白試驗采用0.2g 蔗糖代替煤樣,試驗步驟與煤樣分析相同。
注:每日在煤樣分析前,冷凝管須用蒸汽進行沖洗,待餾出物體積達100~200mL 后,再做正式煤樣。
4.5 分析結果的計算


4.6 氮測定的精密度
氮測定的重復性和再現性如表2 規定:
5 氧的計算

6 結果換算
按下列公式可將空氣干燥基的碳、氫、氮、硫、氧含量換算成收到基、干燥基和干燥無灰基的含量:
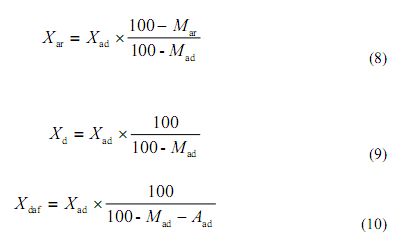
.當這空氣干燥煤樣中碳酸鹽二氧化碳含量大于2%時,則

式中X 代表碳、氫、氮、硫或含量,% 。
_______________________
附加說明:
本標準由中華人民共和國能源部提出。
本標準由煤碳科學研究院北京煤化學研究所歸口并與云南煤田地質勘探公司143 隊共同起草。
本標準主要起草人郭綱才、王廣育、馬尊美。
本標準由于1964 年首次發布。
本標準委煤炭科學研究總院北京煤化學研究所負責解釋。




